Non-coding RNAs
and Gene Therapy
A
significant number of diseases involve a gain of function. This gain of
function is either due to a mutant gene, or due to loss of regulation of a
gene. In addition, there are also diseases that require a specific component of
the cellular machinery to propagate. In all of these cases, silencing a gene
that is either itself responsible for the disease or is involved in its
propagation, can serve to alleviate the disease symptoms. RNA interference is a
naturally occurring phenomenon that the cells employ in order to regulate gene
expression during development as well as in routine metabolism. This mechanism
involves the use of anti-sense RNA to recognize the target mRNA, which is
subsequently degraded. Recently, researchers developed gene-silencing
approaches based on RNA interference by designing synthetic small interfering
RNAs (siRNA), as well as DNA constructs that get transcribed to short hairpin
RNAs (shRNA) in the cell. The shRNAs are in turn processed by cellular enzymes
to siRNAs. RNA interference through shRNAs has proved to be an effective and
long term silencing mechanism that can potentially be applied to many human
disorders ranging from cancer and prion diseases to the diseases caused by
viruses such as HIV & HPV.
Although the classical view of a gene was that of a DNA sequence that gets transcribed and subsequently translated into a protein, a huge chunk of the eukaryotic genome, in fact even larger than the protein-coding part, is transcribed into non-coding RNAs (1). Recent studies on mouse transcriptome have revealed that ~72% of all the transcripts overlap with those transcribed from the opposite strand. Some of these overlapping transcripts play a very important role in gene regulation. In a naturally occurring gene regulation mechanism, small oligo-RNAs, 20-22 nucleotides (nt) in length, act as potent gene silencers leading to the sequence specific degradation of mRNAs with sequences complementary to them. These silencing (antisense) oligos are produced as a result of the cleavage of double-stranded RNA by an RNase III family enzyme called Dicer, and the subsequent binding and melting of the double-stranded cleavage products by a multi-protein complex called RISC (RNA-induced silencing complex) that degrades the target mRNA. The double stranded fragments generated by Dicer are ~22 bp in length and are known as small interfering RNAs (siRNA) and the phenomenon is known as RNA interference (2, 3, 6). The strand that is complementary to the target mRNA is called the guide (as it guides the RISC to the taaret mRNA), whereas the strand complementary to it is known as the passenger.
The siRNAs have certain particular characteristics that are critical to their function. In addition to their characteristic size, siRNAs always have a 2nt overhang at 3’ end of the potential guide strand. The double-stranded (dsRNA) precursor of siRNA can either be from an infecting virus, or part of an organism’s own regulatory machinery. The endogenous dsRNAs are hairpin (stem-loop) structures encoded as transcripts that are 5’capped and polyadenylated and may be up to thousands of nucleotides long, called primary microRNAs or pri-miRNAs (figure-1). The pri-miRNAs are cleaved within the nucleus by an RNase III family enzyme called Drosha, which cuts ~ 22bp away from the loop of the stem-loop structure (now called a pre-miRNA). However, drosha leaves a 2nt overhang on the 3’ arm (would be guide) of the hairpin. This overhang serves as a nuclear export signal in an Exportin5/Ran-GTP mediated export. Once in the cytoplasm, the pre-miRNA is cleaved by Dicer ~22bp from the base of stem-loop, generating the siRNA. The siRNA is then loaded onto RISC, the passenger strand expelled & degraded, and the guide used to target and subsequently cleave the target mRNA.
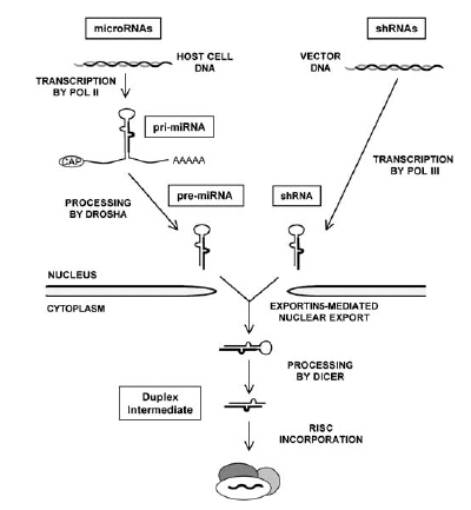
Figure-1: an overview of the processing events, starting either from microRNA transcription or from an artificial hairpin construct, both of which in the end converge on the generation of 20-mer double-stranded siRNAs. (taken from B R Cullen 2006)
Based on what occurs naturally in the cell, it is possible to design artificial RNA interference tools (2, 3, 4, 6). There are two approaches to this technique; one focusing on producing artificial siRNAs, while the other focusing on generating siRNAs in-situ by transforming cells with DNA constructs coding for hairpin-RNAs closely resembling their natural orthologs in structure and function. Among the caveats to making artificial RNAs are that they degrade very fast in a living cell owing to the cellular cocktail of different nucleases. Some studies and projects actually managed to get around this problem by chemically modifying the siRNA backbone (5). It was found that chemically modifying the 2’ position of the sugar in the backbone greatly enhances the efficacy of synthetic siRNA, however, there still remains the issue of dosage since it requires very high titers of the RNA drug to knockdown the problem-gene. Nonetheless there is still intensive research underway, with many successes, to increase the silencing potency of synthetic siRNAs (2, 4). However, synthetic siRNAs can hardly match the cost efficiency, length of expression, dosage multiplicity and the lack of adverse effects offered by in situ production of siRNAs. This is achieved by using hairpin-encoding DNA expression cassettes inserted into either plasmid or viral vectors; or in some cases even into dumbbell-shaped, dual-hairpin DNA constructs, found to be resistant to exonuclease attack (4). In-situ production of siRNAs bypasses the need to administer synthetic siRNAs in large doses. Moreover, it also compensates, through sustained production of siRNAs, for the inevitable enzymatic breakdown and turnover as long as the DNA construct stays intact.
Despite the versatility and the potential for long-term expression, there are certain crucial properties of natural siRNAs, which if not taken into consideration in designing the hairpin-cassettes, can render the constructs ineffective. The first property to be considered is the 2nt overhang at the end of the 3’ arm of the hairpin (prospective guide). This is where the Dicer grabs onto when cleaving the hairpin to yield a siRNA (6). The size of the loop has also been found to be important, it is desirable to have a loop size ~9nt (3). Loop sequence is also important, using a loop sequence from one of the natural microRNAs yields better results, even with loop sizes of 4-7nt. A very important factor is the relative stability of the 5’ ends of the two complementary strands of a siRNA; in fact this is what determines which of the two will become the guide strand. When a siRNA is loaded onto the RISC, almost always the strand with the more loosely bound 5’ end becomes the guide while the one with relatively strongly base-paired 5’ end becomes the passenger and is therefore expelled and degraded. Hence having A-U base-pairs at the loop-closure is helpful, in fact having a G-U mismatch in the proximity is even better (6). On the other hand, it is good to have two G-C base-pairs at the base of the stem-loop in order to make the 5’ end of the passenger strand more tightly bound (6). In general, the hairpin should be as low in G-C content as possible so as to allow easy melting; it is also desirable to have a few mismatch-bulges in the stem (figure-2).
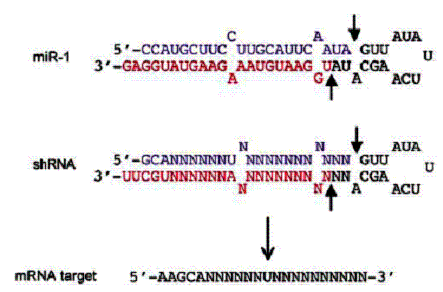
Figure-2: A proposed blueprint of an effective short
hairpin shRNA design based on the important sequence landmarks of a naturally
occurring microRNA, miR-1. The arrows indicate the dicer cleavage sites. (taken
from B R Cullen 2006)
It is also important to have a high level of expression of the hairpin expression cassette inside the treated cells therefore usually pol III promoters are used for expression, which can sustain a higher level of transcription than pol II promoters. In addition, using pol III promoters also rules out the risk of competing with the cell’s housekeeping genes for the polymerase. Moreover, the inherent nature of pol III termination mechanism adds ~2 Ts, 3’ to all transcripts, which in a hairpin can serve as the required overhang (for dicer) at the end of 3’ arm (6). In addition to efficient melting and proper RISC loading, it is also necessary to choose a target sequence available and easily accessible for the guide-RISC complex to bind. First, the chosen sequence must be present in the target spliced variant (or variants) of the mRNA, and second, it should be made sure that the target sequence is not sequestered into a secondary structure, in which case it will not let the guide-strand properly bind to it thereby rendering the guide-strand ineffective. Bioinformatics has provided much help in this regard and these days there are a number of web-based programs that can rule out bad sequence choices in shRNA design.
Another major issue in ‘RNAi-based Gene Therapy’, and an extremely important one is the choice of a vehicle for the delivery of the shRNA cassette. In this area there is a huge diversity of approaches ranging from simply injecting naked-plasmids into blood stream to using highly sophisticated, custom-designed, viral and bacterial vehicles; some of which can even integrate the their ‘payload’ into the host cell genome (2).
Retroviruses are a popular choice among gene therapy vehicles, not just because of the ease of production and modularity of components (i.e. the components can be mixed and matched), but also due to the versatility of tropism (targeting) and in some cases their ability to integrate into the host genome. These are RNA based viruses that undergo reverse transcription inside the host cell, using the enzymes that they carry in their viral capsids. This process consists of an elaborate series of steps involving DNA synthesis using the RNA template of the viral genome, and strand transfers followed by degradation of the RNA template. The end result is a DNA provirus with long terminal repeats (LTRs) at both ends, which contain the promoter and enhancer elements (U3 of 5’ LTR) for the viral genome as well as sequences required for integration (figures-3, 4).
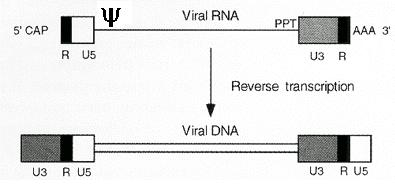
Figure-3: Retroviral genome before and after reverse transcription. (taken from J.M. Cofflin, et al.1997)
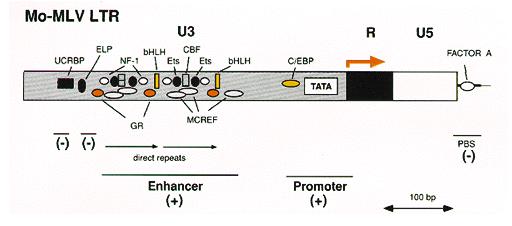
Figure-4: Enlarged view of the 5’ LTR showing the U3 enhancer and promoter elements and the binding sites for various transcription factors. (taken from J.M. Cofflin, et al.1997)
However packaging and purification
are the issues that need to be carefully considered if the technique is to be
extended beyond experimental animals and to be applied to humans. A retroviral
particle can only be produced by a eukaryotic cell (a packaging cell, derived
from a rodent, primate or human cell line) that expresses the genes encoding
all the viral components required for viral packaging (capsid proteins),
infection (envelope glycoproteins), and reverse-transcription & integration
(reverse-transcriptase/integrase) into the host genome. These genes however, must
not be present in the therapeutic-payload that the virus carries; otherwise,
the payload-construct will produce a second generation of infectious viruses
from within the transduced cells. Therefore, it is very crucial that the
payload-construct, a therapeutic virus carries, is replication-defective.
This is achieved by deleting all the genes coding for the viral proteins from
the viral genome; in addition, the promoter elements (U3) that drive the
expression of the virus inside the host are also deleted, and only the
sequences in the original genome that are required for proper packaging of the
virus (y-sequence,
see figure-5) and for reverse transcription inside the transduced cell are
left. The hairpin (shRNA) expression cassettes are then inserted into these
viral backbones that have their own promoters. Hence the only part of the
recombinant-therapeutic virus that is capable of being transcribed in the host
is the hairpin expression insert it carries. The viral proteins required to
make a viral vehicle are expressed in the packaging cells in-trans, from
separate constructs (packaging-constructs). Thus although the therapeutic viral
vehicle is made up of the building-blocks of a wild type virus (hence the same
target specificity), it cannot cause an infection because it does not express
any viral proteins itself (figure-5).

Figure-5: A recombinant, therapeutic retroviral construct
with GFP as a reporter-gene, note the shRNA sequence under the H1 promoter, and
the lack of viral-genes (region between y& PPT
deleted), which are provided to the packaging cell in-trans. (taken from A.
Pfeifer et. al. 2006)
However, the retroviral packaging is not that simple, it is very difficult to produce vectors in high titers because often the viral genes are toxic to the packaging cells & certain mechanisms in the packaging cells retard proper expression of viral genes.
It is also possible for therapeutic-construct to undergo recombination with either the packaging-construct (that codes for viral proteins), or with a wild-type virus of either the same or similar form that might be present in packaging cell line as an integrated, dormant provirus. In addition, the packaging construct may also serve to ‘pack’ an integrated, replication-defective virus besides the therapeutic-virus, causing contamination. One solution to these problems is to break the packaging construct up into smaller units, each expressing a single gene, and to integrate those units in different places in the packaging cell genome. In addition, the contaminating viruses can be gotten rid off through CsCl gradient centrifugation (8). The issue of packaging and purification is still a ‘hot spot’ in the field of gene therapy.
The issues concerning effective retroviral gene therapy are not just limited to design and packaging, some very severe problems can arise only after the ‘right’ construct has been delivered to the ‘right’ cell. The capability of a retroviral-vector to integrate in the host genome can be both good and bad depending on where it integrates, because if it integrates in the middle of a gene or a regulatory sequence, or a splice site etc., it can have devastating effects, ranging from genes shut off to genes going out of control. Thus curing one disease may end up causing another, or even cancer due to insertional mutagenesis.
Most retroviruses can only transduce dividing cells since their capsids cannot pass through the nuclear pore complex, thus can only transduce a cell during mitosis, when the nuclear envelope disintegrates. However, lentiviruses, which are a subtype of retroviruses, are capable of even transducing fully differentiated, non-dividing primary cells. Their capsid proteins are distinct from other retroviral capsids in their ability to interact with the nuclear pore complex, and hence get actively transported to the nucleus. Therefore lentiviruses are the most preferred vehicle when transducing non-dividing, primary cells such as neurons.
Adenoviruses are another important viral vehicle used in gene therapy. These are non-enveloped, double-stranded DNA viruses. The virions enter the host cell through receptor-mediated endocytosis. The DNA ultimately makes its way to the nucleus, while the capsid and coat disintegrate on the way. Once inside the nucleus, the viral genome exists as episomal DNA and expresses two families of genes: the early or E genes that encode proteins required for replication, and the late or L genes that encode proteins required for packaging. The viral vectors commonly used have many of their E genes deleted and most of L genes removed, which are provided in trans in the packaging cell. However, the packaging sequences (ITRs etc.) are retained. To generate recombinant viral vectors the shRNA construct is first cloned into a shuttle vector from which it is transferred to the circularized viral genome either by homologous recombination between the viral and shuttle plasmids, or by site-specific transposition into viral plasmid.
The recombinant
viral plasmid is then linearized and transfected into a packaging cell, which
packages it into infectious virions (8). Adenoviruses are the vehicle of choice
when integration is not desired. Adenoviral vectors are sometimes used to
express a ‘second generation’ of retroviral vectors that are
replication-defective. In this case, the primary vector (adenoviral) expresses
the secondary vector (retroviral), thus turning the transduced cells into a set
of in-vivo-packaging-cells. This technique enhances the efficacy of the
retroviral vectors, first by providing an additional amplification step, and
second by facilitating their generation & release close to their target
sites (8, 10).
A recent study (7) used lentivector-mediated RNAi to suppress the cellular prion PrPc protein and thus to make the cells refractory to the exposure to Encephalopathy-causing prion, PrPSc (7). The group first designed hairpin constructs, using open-access web programs against 6 different sites on the Prnp mRNA that translates into PrPc. The viral constructs were then used to transduce N2a neuroblastoma cells. One construct named LVsh512, covering 512-532nt of the mRNA, was found to be more than 90% efficient. This estimate of efficiency was based on the expression of the reporter-construct (EGFP driven by PGK promoter), and on western blots that confirmed the suppression of PrPc in the transduced cells in culture (figure-6).
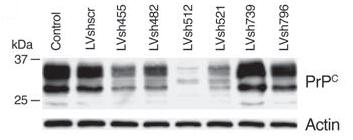
Figure-6: A western blot (72h post-infection) showing
PrPc levels in cells transfected with different hairpin silencing lentivectors,
with uninfected cells as the control. Note the significant reduction in the
target protein levels in cells transfected with LVsh512. Actin
here is used as the loading control. (taken from A. Pfeifer et. al. 2006)
Later, in order to
test the efficacy of the silencing-lentivector transduction in primary
non-dividing cells, the group infected cerebellar granule cells with LVsh512.
As a control, a set of granule cells was infected with LVEGFP, the viral vector
only expressing GFP and not the shRNA. As before, LVsh512 significantly
silenced PrPc expression; on the other hand, LVEGFP caused no significant
decrease in PrPc levels despite expressing GFP at levels comparable to LVsh512.
This showed that PrPc suppression was not an artifact of viral infection
(figure-7).
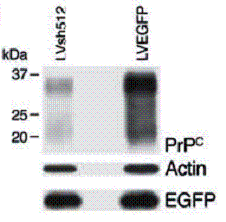
Figure-7: A western blot
(72h post-infection) showing PrPc and GFP levels in cerebellar granule cells
infected with either LVsh512 or LVEGFP (also see figure-5 above). (taken from
A. Pfeifer et. al. 2006)
The study was
extended further by transfecting the silencing lentivector (LVsh512) into a
cell line (ScN2a) that had chronically been exposed to the disease-causing form
of cellular prion protein, PrPSc (prion protein scrapie). When a cell is
exposed to the mutant, disease-causing prion (PrPSc), the cellular prions
(PrPc) also acquire the characteristic, misfolded 3D structure of the PrPSc
that is protease resistant. Accumulation of misfolded prions in nerve cells
results in neuronal dysfunction and cell death, leaving behind empty spaces and
hence the name spongiform encephalopathy. Thus, although the cellular prions
(PrPc) take the same misfolded appearance after being exposed to the scrapie
prion (PrPSc), they are not identical to the scrapie prion in sequence. In the above-mentioned experiment control
ScN2a cells were infected with LVshscr, expressing shRNAs against the mutant,
scrapie prion. The results (western blots using antibody against scrapie
isoform) indicated expression of protease resistant scrapie-prion in the
uninfected cells and in cells infected with LVshscr, but almost no
scrapie-prions were detected in the ScN2a cells infected with LVsh512. This
proved that the scrapie-prions accumulating in the ScN2a calls were the
cellular prions (PrPc) that had been misfolded into PrPSc isoform, and not the
mutant-prions. Detection of scrapie-prions in LVshscr-infected cells indicated
that knocking down the mutant-prion gene does not affect the accumulation of
scrapie-isoform, unless the cellular prion gene is knocked down, as in case of
LVsh512 (figure-8).
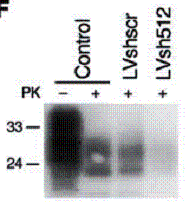
Figure-8: A western blot (72h post-infection), indicating
the presence of scrapie isoform of prion in the uninfected cells (control),
LVshscr-infected cells and in cells infected with LVsh512. The + and – signs
above the lanes refer to the presence or absence of protease-k. (taken from A.
Pfeifer et. al. 2006)
In order to test the efficiency of the lentivector
silencing system (LVsh512) inside a living animal, the viral vectors were used to
transfect the brain cells of homozygous tga20 mice, through intracranial,
stereotactic injections. These mice have 20 copies of the prion gene and
express up to 10 times the normal levels of the cellular prion protein. As a
control, a set of mice was infected in the same way with the silencing
lentivector against the mutant prion (LVshscr). The results indicated
comparable levels of GFP expression in both the experimental and control
animals, however, only the mice infected with LVsh512 showed significantly low
levels of cellular prion expression in the infected tissues.
Finally, the silencing lentivectors (LVsh512) were used
to transfect 129Sv-derived embryonic stem cells (ES), and the infected ES cells
were injected into WT C57BL blastulae to generate chimeric mice. The degree of
chimerism in the offspring was verified by the extent of agouti color (a
phenotypic marker of 129Sv line) in the coat of chimeras. These estimates of
chimerism were also verified by immunohistochemistry and RTPCR. In a 90% chimeric
mouse, GFP (the reporter for shRNA) was found to be expressed over most of the
brain as revealed by florescence imaging (figure-9).
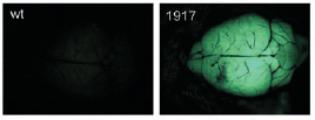
Figure-9: GFP expressed
as a reporter for shRNA expression in a chimera (1917). Note the lack of GFP in
the wild type control specimen on the left. (taken from A. Pfeifer et. al.
2006)
Furthermore, as an extension of the same experiment, a set of chimeric mice was intra-cerebrally inoculated with the misfolded scrapie prion in an attempt to expose them to scrapie. The same procedure was also applied to three sets of control mice, WT 129Sv, GFP.3 (infected with LVEGFP lentivector), and 129Sv ´ C57BL hybrids. The average lifespans of control lines were found to be approximately 167, 168 and 163 days post-infection for WT 129Sv, GFP.3 and 129Sv ´ C57BL hybrids respectively. The average lifespan of less than 35% chimeras was ~179 days, not significantly more than the controls; however, the average lifespan of more than 65% chimeras was ~214 days, which was significantly more than the controls. A ~95% chimeric mouse actually survived for up to 231 days post infection.
The above-described study quite clearly highlights the effectiveness of shRNAs in silencing in silencing cellular components that might serve to propagate a disease, such as spongiform encephalopathy incase of prions. Of great importance was the approach of employing the RNAi against cellular prions in cells that had already been exposed to disease causing scrapie. This proved that if the gene that helped scrapie to propagate could be silenced, accumulation of the scrapie prion and its associated symptoms could be prevented, at least in a culture dish. However, no one knows what might be the consequences of silencing a gene such as cellular prion, especially in humans and in the long run. However, as seen with mice, it might serve to lessen the severity of the disease and prolong the lifespan. Many experimental approaches involving gene therapy seem to work perfectly well in mice mainly because of the short lifespan of the subjects and due to the short duration of the study. As in case of prion silencing (7), the authors either did not encounter or did not mention any instances of insertional mutagenesis despite the long-term expression that was quite likely due to the vector integrating into the host genome. One explanation could be that there simply was not enough time allowed for some of the adverse effects of integration to elicit. Nonetheless, it’s hard to assess if a technique can be safely applied to human subjects, just by considering the results of short-term experiments on mice. There is still a lot of room for research in this area specially the research that focuses on the long-term adverse effects and complications, not just of integrating a chunk of DNA into the host genome, but also of silencing a host gene in the long run.
The research so far in the field of silencing gene therapy has been quite promising and holds a lot of potential especially in treating diseases that are caused due to a gain of function such as many cancers and tumaors. In addition, RNAi can also be directed against pathogens themselves, especially retroviruses such as HIV (11), however these techniques are still in their infancy and a huge challenge facing this approach is the fast mutation rate of viruses like HIV. All to often the virus against which the RNAi is directed mutates, rendering the silencing system ineffective. One solution to these escape variants is to design long hairpin RNAs (lhRNA) rather than shRNAs. The lhRNAs get processed by Dicer into multiple siRNAs, each directed against a different site on the target. There is still research underway to eliminate the caveats of this technique, which one day may become one of the most preferred RNAi approaches.
One issue concerning RNAi that was mentioned in the very beginning is the property of the siRNA to load efficiently onto RISC (5), which is considered to be a rate-limiting step and is also a direct measure of the silencing capability of the siRNA (5, 6). One study employed an innovative approach to testing the RISC binding ability of a siRNA. The technique involved co-transfecting a fixed concentration of a standard siRNA, directed against a reporter gene whose expression could be easily quantitated (e.g. GFP etc.), into the same cell as the siRNA whose effectiveness was being measured. It was theorized that the test-siRNA would compete for RISC binding with the standard-siRNA, thereby decreasing the extent of reporter gene suppression. In other words, the effectiveness of the test-siRNA would be correlated to the increase in the expression of the reporter gene it causes; this increase in the reporter gene expression can be easily quantitated thus providing a measure of the effectiveness of the test-siRNA. This technique, if refined and further developed, can serve as an effective assay to measure the effectiveness of an shRNA and can revolutionize shRNA design.
REFERENCES:
1) Mattick JS, Makunin IV. Non-coding RNA.
Hum Mol Genet.
2006 Apr 15;15 Spec No 1:R17-29.
2) Racz Z, Hamar P. Can siRNA technology provide the
tools for gene therapy of the future? Curr Med Chem. 2006;13(19):2299-307.
3) Inoue A, Sawata SY, Taira K. Molecular design and delivery of
siRNA.
J Drug Target.
2006;14(7):448-55.
4) Shiota M, Ikeda Y, Wadhwa R.The factors that contribute to the long-term expression of
siRNA.
Nucleic Acids
Symp Ser (Oxf). 2006;(50):243-4.
5) Koller E, Propp S, Murray H, Lima W, Bhat B, Prakash TP,
Allerson CR, Swayze EE, Marcusson EG, Dean NM.Competition for RISC binding predicts in vitro potency of
siRNA. Nucleic Acids Res.
2006;34(16):4467-76. Epub 2006 Aug 31.
6) Cullen
BR.Induction of stable RNA interference in mammalian cells.
Gene Ther. 2006 Mar;13(6):503-8.
7) Pfeifer
A, Eigenbrod S, Al-Khadra S, Hofmann A, Mitteregger G, Moser M, Bertsch U,
Kretzschmar H.Lentivector-mediated RNAi efficiently suppresses prion
protein and prolongs survival of scrapie-infected mice.
J Clin Invest. 2006
Dec;116(12):3204-10.
8) Templeton, NA. Gene and Cell Therapy:
Therapeutic Mechanisms and Strategies. Marcel Dekker Inc. New York. 2004:10-60.
9) Coffin, John M.; Hughes, Stephen H.; Varmus, Harold E.
Retroviruses.
Plainview (NY): Cold
Spring Harbor Laboratory Press. 1997: Chapter 4.
10) Okada
T, Caplen NJ, Ramsey WJ, Onodera M, Shimazaki K, Nomoto T, Ajalli R, Wildner O,
Morris J, Kume A, Hamada H, Blaese RM, Ozawa K. In situ generation
of pseudotyped retroviral progeny by adenovirus-mediated transduction of tumor
cells enhances the killing effect of HSV-tk suicide gene therapy in vitro and
in vivo.
J Gene Med. 2004
Mar;6(3):288-99.
