Deletion and Duplication of Chromosome 7q11.23
Williams syndrome (WS; OMIM 194050) and 7q11.23 duplication syndrome (Dup7; OMIM 609757) are two rare neurodevelopmental disorders caused by deletion and duplication respectively, of 25 genes on human chromosome 7. They present with an array of neuropsychiatric disorders, such as autism spectrum disorder (ASD), anxiety, attention deficit, depression, and intellectual disability, making them useful for studying the molecular mechanisms of common mental health disorders. Understanding the molecular basis for these syndromes will allow the development of novel, directed therapies for their debilitating features, and also offer insight into normal brain development and function.
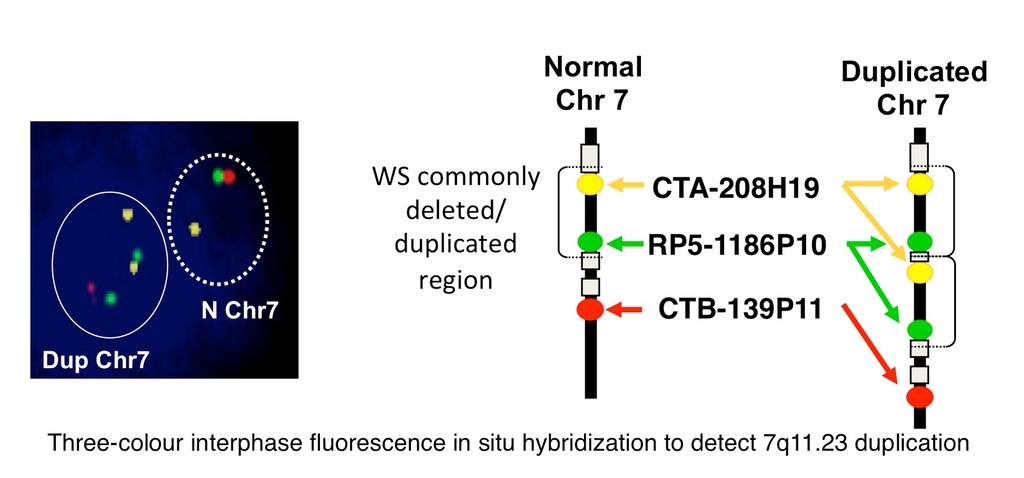
The Osborne lab studies these two disorders using both human participants and animal models. We have helped characterize the structure of the 7q11.23 genomic region and to unravel the mechanisms behind the 1.5 million base pair deletion, through the identification of a common inversion polymorphism that increases the risk of deletion or duplication of 7q11.23 in offspring.
In 2005 we published the first case of a child with Dup7, so providing a novel entry point towards finding new genes required for normal speech and expressive language, which are impaired in this group of children.
In particular, our lab studies the roles of two proteins, GTF2I and GTF2IRD1, which are likely responsible for many of the features of WS and Dup7. We have generated mouse models with deletion or duplication of these two genes as well as induced pluripotent stem cell lines from individuals with WS or Dup7 and are using these resources to investigate the neurobiological bases for these disorders.
Epigenetics of WS and Dup7
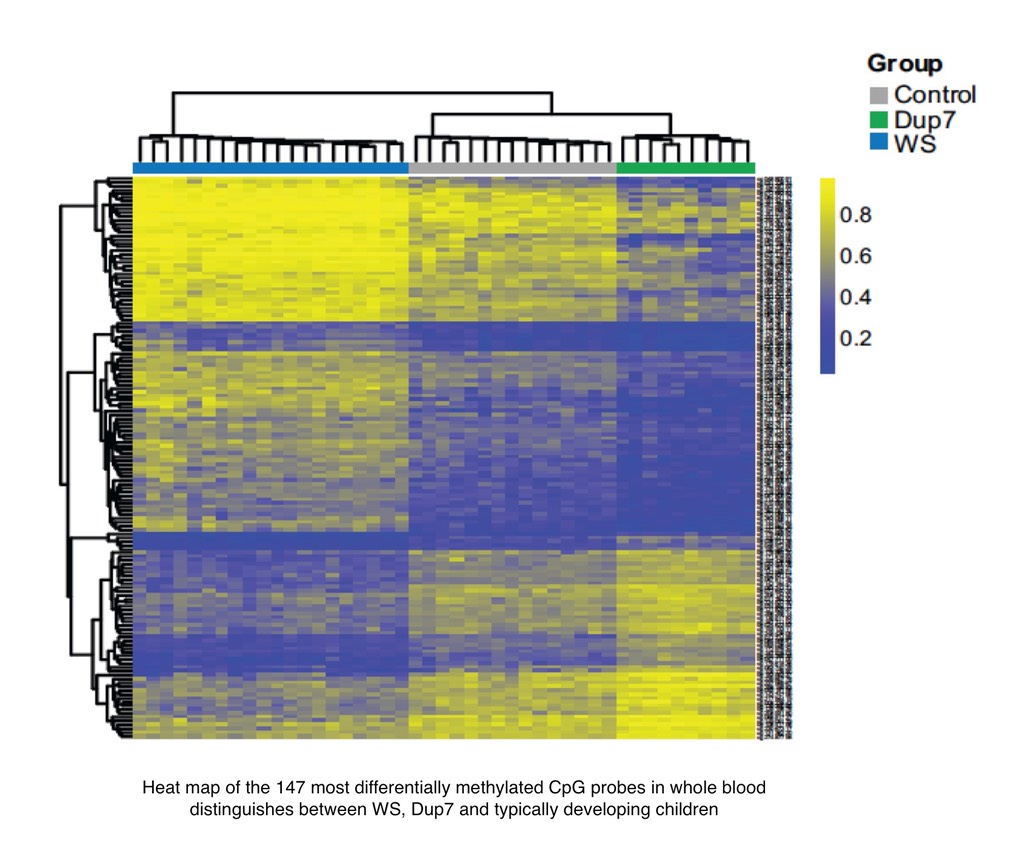
We recently identified genome-wide, symmetrical differences in DNA methylation in blood DNA from children with these two syndromes, the majority of which were associated with genes involved in brain development, function and disease. We are now building a multi-layered epigenetic map of WS and Dup7 in tissues more relevant to the cognitive and behavioural symptoms of these disorders, using patient-derived stem cells and primary cells from mouse models. With parallel approaches in humans and mice, differentially expressed genes will be cross-referenced with DNA methylation, ChIP-seq and HiC data to identify biologically relevant epigenetic changes associated with 7q11.23 gene deletion and duplication.
Genotype-Phenotype Associations in WS and Dup7
In collaboration with Carolyn Mevis (University of Louisville, KY) and Colleen Morris (University of Nevada, Las Vegas) we recently published psychological and clinical descriptions of a large group of children and adults with Dup7. Common diagnoses included separation anxiety, social and specific phobias, selective mutism, attention deficit hyperactivity disorder and oppositional defiant disorder. We hope to correlate genetic and epigenetic data with the in-depth clinical and psychological data collected for both our WS and DUp7 cohorts.

We have also carried out the first systematic characterization of autism spectrum disorder (ASD) symptomatology in Dup7 which found a clinical diagnosis for 20% of the children studied. We hypothesized that these children who are carriers of the 1.5 Mb 7q11.23 duplication and that have a diagnosis of ASD will carry additional rare damaging variants in ASD-relevant genes, and those that do not have an ASD diagnosis will not. We are currently performing whole genome sequencing (WGS) analysis of children with Dup7 and ASD (Dup7-ASD) and children with Dup7 and no ASD diagnosis (Dup7-non-ASD) in collaboration with Ryan Yuen (SickKids). We will compare the burden of potentially pathogenic rare variants in these two groups to determine whether ASD group carry a significantly higher burden of rare variants that accounts for their diagnosis.
Neurobiology of 7q11.23 Disorders
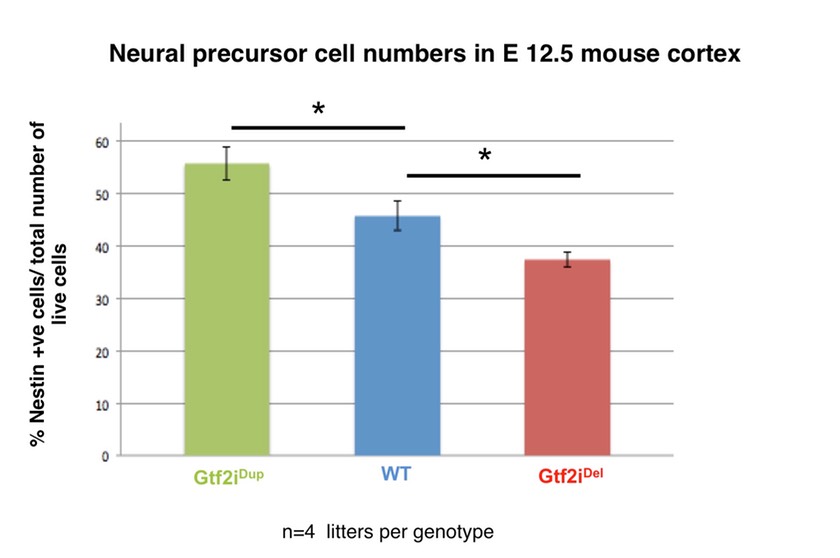
We have identified reciprocal defects in neuronal precursor growth and proliferation, cell fate and cortical organization associated with deletion and duplication of Gtf2i. With our collaborator, Zhong-Ping Feng (Physiology, UofT), we have found reciprocal defects in axon branching and calcium influx in primary cortical neurons from these same mouse models that are directly caused by alterations in the cellular localization of the TRPC3 calcium channel.
In collaboration with James Ellis (SIckKids) we have carried out genome-wide expression and functional analysis in neurons grown from WS patient-derived induced pluripotent stem cells, and found changes associated with the WS deletion, giving the first indication of possible pathways involved in brain development and/or function in this population. We are now expanding these studies to understand how changes in cellular function and gene expression drive the altered neuronal development and maturation in mouse and human.