Pulmonary Arterial Hypertension (PAH)
Pathophysiology: Any process that effectively leads to an
increased intrapulmonary vascular resistance can cause PAH.
Traditionally, etiologies divided into capillary, pre-capillary,
and post-capillary causes.
Classification:
(1)
Idiopathic or Primary Pulmonary Hypertension: no known cause
(2)
Secondary Pulmonary Hypertension: Etiologies include:
precapillary causes: pulmonary vasculitis, collagen vascular
diseases, multiple pulmonary emboli; rarely, multiple vascular
tumor emboli, pulmonary endarteritis obliterans often aggravated
by underlying PAH, marked emphysema or bullous emphysema which
may attenuate the smaller pulmonary arteries, post-surgical or
radiation elimination of sufficient pulmonary arterial bed,
drugs
capillary causes: any diffuse, infiltrative lung disease
involving numerous alveolar septae
post-capillary causes: valvular heart disease especially
mitral stenosis, chronic heart disease, left atrial myxomas,
tumors or blood clots
Commonest Causes: severe COLD/emphysema, sleep apnea
Clinical Clues: Progressive dyspnea, decreasing exercise
tolerance, tachypnea and tachycardia, right ventricular strain
on ECG, congestive heart failure and cor pulmonale.
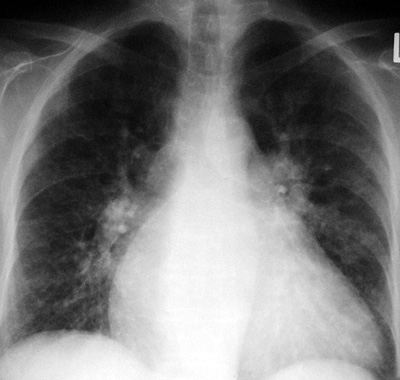
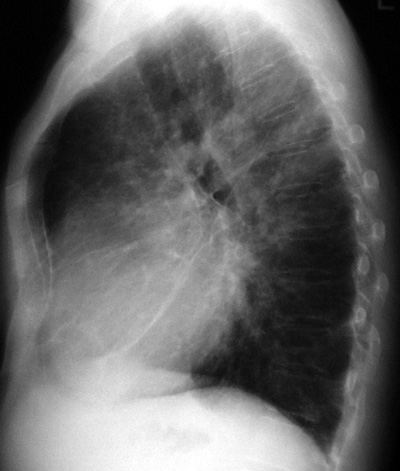
CXR Findings:
1.
prominent central pulmonary arteries ie. big hila
2.
thinning and attenuation of pulmonary arterial branches
3.
abrupt cut-off or squaring of pulmonary arteries
4.
increase in vascular-free peripheral lung zone
5.
right ventricular dilatation and cardiomegaly
6.
any underlying lung pathology that may be a cause
“Aunt Sophies”: the main mimickers are other causes of
large central hila
1.
Hilar adenopathy of any cause **
2.
Idiopathic dilatation of the pulmonary arteries, seen in
asymptomatic young women; this is only a CXR problem, not a
physiologic one.
|