TOP
Retinitis Pigmentosa
NOTE 1: If you are unfamiliar with the anatomy of the eye, the structure of the retina and the functioning of the visual pathway, please visit: The Visual Pathway
NOTE 2 : In most cases, clicking on an image will enlarge that image thus showing more of its details
Introduction
Clinical Diagnosis
Modes of Inheritance
The Fundus of the Eye
Histological Studies
The Molecular Pathology of Retinitis Pigmentosa
Therapy
Conclusions
Acknowledgements
References
Retinitis pigmentosa (RP) refers to a group of diseases that
affect about 1.5 million or one in four thousand people around
the world. Retinitis pigmentosa, as the name implies, is a disease of the retina but
as we will see later it specifically affects the retinal rod
photoreceptor cells.
Patients with RP do not show any systemic, metabolic, inflammatory, toxic, traumatic
or dietary condition.
(1.). However, 5% of patients have profound congenital deafness and a vestibular
disorder (Usher syndrome type I), and 15% of patients have associated partial
hearing loss (Usher syndrome type II or type III), and some rare forms of retinitis
pigmentosa are associated with systemic neurological disease. These are:
- Bassen-Kornzweig disease,
- Friedreich-like ataxia with retinitis pigmentosa and
- classic Refsum's disease, which are associated with systemic neurological disorders. (1a.)
In the past ophthalmologists believed that RP was caused by an
inflamation of the retina, hence the name, Retinitis (compare
pneumonitis in the reference to the inflamation of the lungs or
peritonitis in thereference to the inflammation of the peritoneum,
the lining of the abdominal cavity).
The word "pigmentosa" comes from the observations of
the early ophthalmologists that black pigment could be seen on
the retinas of RP patients when they looked into their eyes with
their ophthalmoscopes (see Figure 5).
Although pigment does appear on the retinas of RP patients,
the reference to retinitis as an indication of inflammation turned out to be a misnomer. The
use of light microscopy, the advent of high resolution microscopy
and, more importantly, the development of sophisticated
biochemical and genetic techniques, led to the conclusion that the disease is
due to degenerative changes within the visual cells of the retina,
(see below).
Back to TOP
Clinically, symptoms of retinitis pigmentosa include:(2.)
- impaired dark and light adaptation, progressive night blindness, and
difficulty with mid peripheral visual field in the early stages of the disease.
- A progressive decrease, and eventual loss, of the electroretinogram (ERG), a measure of the electrical response of the retina to a flash of light.(see Figure 1)
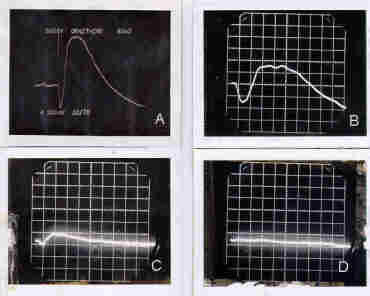
Figure 1 - Electroretinograms of:
- a normal retina {(A) (upper left)} . Note the slight decrease in the electrical response to the flash of light (the a-wave) and the sharp rise in the amplitude of the b-wave which falls sharply to ground level.
- an electoretinogram of a retina showing early development of RP {(B) (Upper right)}. Note that the amplitudes of both the a-wave and the b-wave have decreased.
- an electrogram of a retina in the final stages of retinal degeneration due to RP{ (C) [Lower left]}. Note that the amplitudes of both the a-wave and the b-wave have become markedly reduced.
- an electoretinogram of a retina that has lost its response to light due to degeneration of the photoreceptor cells as a result of RP{(D)[Lower right]}. The patient would now be blind.
Usually, both eyes are affected by RP. In some cases, however, only one eye may be affected while the other eye remains normal (see Figure 2).
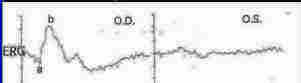
Figure 2 - Chart showing the ERG's of a normal (OD, left panel) and an RP eye (OS, right panel) from the same individual. Note that the ERG in the left panel looks normal while the one to the right is completely extinguished. (Modified from a slide provided by Dr. S. Coupland, Ottawa Eye Institute, Ottawa University, Ottawa, Ontario)
- Constriction and gradual loss of the visual fields
- Later, loss of peripheral fields and "gun barrel"
vision
- Loss of visual acuity
- Appearance of pigment granules or "bone spicules" on the surface of the retina
- Attenuated retinal vessels
- Waxy Palor of the Optic Disc
- a tendancy towards color blindness
- Cataracts develop in most cases, and some have cystoid macular edema.
- eventually loss of central vision and blindness.
Back to TOP
Retinitis pigmentosa can be inherited as an autosomal dominant,
as an autosomal recessive, or as an X-linked disease. In the X-linked disease,
mothers are carriers and the males only of the family are
affected while the female become carriers.
Some patients show RP only in one eye (see Figure 2), while in others Retinitis pigmentosa is associated with other diseases as part of
a syndrome. In the vast majority of cases, the isolated or sporadic cases, a genetic
link cannot be determined.
It is difficult to identify the relative frequency of different types of RP. The largest single group is the isolated or sporadic patients, usually comprising 40% - 50%
of index cases. These might includes a sizable number of autosomal recessive patients.
Recently, a digenic mode of inheritance for RP was described. In this type of RP a combination of two loci was necessary to produce the clinical picture of RP.(3.)
Oligogenic forms of RP may not be uncommon but they are very hard to demonstrate.
Investigators at the Ottawa Eye Institute report a patient with an affected and a normal eye (Dr. S. Couupland, Personal Communication)(see Figure 2).
Generally, the dominant type of the disease is the most benign form of RP, while the X-linked
type of the disease is the most virulent. Patients with The recessive type of disease start
having problems with their vision by the fifth decade of life.
Back to Top
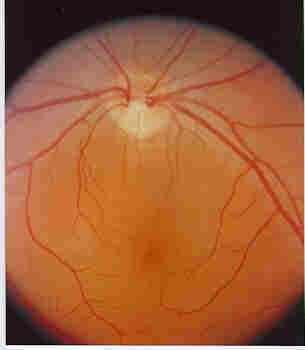
Normally, the fundus shows a uniform pink color with a
Optic disc (yellow disc) through
which the arteries and veins seen on the surface of the retina, enter and leave, respectively
(see Figure 3).
 Click Here to see details of the image
Click Here to see details of the image
Figure 3. - The fundus of a normal Eye. Note the uniform pink color of the fundus and the yellow color of the optic disc. The arteries, which supply nutrition to, and veins, that remove metabolites from, the inner retina, traverse the fundus and enter and exit the eye through the optic disc (Original slide courtsey of Dr. Gilles Desroches, Ottawa, Ontario.
In the early stages of Retinitis pigmentosa, black "bone spicules"
appear on the surface of the retina, the blood vessels become narrower than normal and the optic disc adopts a waxy appearance (Figure 4).
 Click Here to see enlarged image
Click Here to see enlarged image
Figure 4 - The Fundus of a patient with early RP. Note the waxy appearance of the optic disc, the narrowed blood vessels and the black spots ("bone spicules") at the periphery of the photograph. (Original slide courtesy of Dr. Gilles Desroches, Ottawa, Ontario).
As the disease progresses the black spots, increase in number and eventually coalesce (Figure 5) to form blind spots in the
visual fields where the patient cannot see.
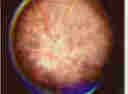 Click Here to see details of the image
Click Here to see details of the image
Figure 5 - The Fundus of an advanced case of an RP retina. Note the numerous accumulations of black spots on the fundus.
In some patients, only one eye is affected while the other eye remains normal (see Figure 2).
Ophthalmologists chart the progress of these coalesced pigment granules
by means of a Visual Field Chart in order to follow the progress
of the patients visual ability.
Back to TOP
Typically the RP patient experiences difficulty seeing under dim light conditions (nyctalopia) as a young adult, followed by progressive loss of both rod and cone vision over the ensuing years to decades.
Immunocytochemical studies using anti-rhodopsin antibodies, revealed shortening of their outer segments of the rods, usually beginning in the mid peripheral
retina and progressing with time to involve the macula and far periphery.
Rod cell degeneration is followed by changes inthe adjoining cones, including
shortening of their outer segments, and ultimately, cone cell
death and blindness.
Additionally, microscopic examination of retinas obtained from human subjects with advanced retinitis pigsmentosa, revealed the absence of rod cells and the presence of ultrastructural abnormalities in both the retinal pigment epithelial cells and the few surviving cone cells. The inner retina appeared to have been unaffected.
The disease in the human retinas examined were was too advanced to allow for the assignment of a primary site for retinitis pigmentosa in the retina. However, the absence of rods in the tissues examined and the fact that the first clinical observation in retinitis pigmentosa is the onset of night blindness supported the suggestion that the disease primarily affects these photoreceptor cells.
This suggestion was enforced by the finding that animals (mice, rats, cats, dogs and pigs) showing electroretinographic changes similar to retinitis pigmentosa, were found to have changes in their photoreceptors similar to those found in RP, namely, degeneration of the photoreceptor cells and eventual loss of the retinal photoreceptor layer (compare Figures 6 and 7).
However, different species of animals exhibited different modes of photoreceptor degeneration. In some, animals degeneration occurred at the time of cell differentiation (some types of mice), others showed changes at the time of the development of the outer segments (Royal College of Surgeons or RCS rats), while still others showed degeneration after the cells have reached full length.
The fact that human subjects with RP continue to have useful vision into their second and later decades of life would suggest that retinal degeneration in these subjects would belong to the second or third categories of degeneration.
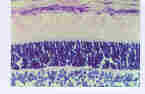 Click here to see details of the image.
Click here to see details of the image.
Figure 6: shows a microscopic picture of a section of a normal retina. The retinal Pigment Epithelium layer, the photoreceptor outer segment layer, the outer nuclear layer, the Outer Plexiform Layer and the inner Nuclear layer are shown.
 Click here to see details of the image.
Click here to see details of the image.
Figure 7 - A degenerated section of an RCS rat retina. Note the complete absence of the photoreceptor layer including the outer nuclear layer.
To understand why black spots appear on the surgace of the retina of RP patients we can take our clue from the findings that the eyes of patients examined showed ultrastructural abnormalities in their pigment epithelial cells. Since we cannot follow the progress of the black spots seen on the fundus of human subjects investigators resorted to the study of animals exhibiting retinal degeneration.
These studies included the protein metabolism in the photoreceptor cells of the so-called Royal College of Surgeons (RCS), which showed that the pigment epithelium plays an important role in the maintenance of the normal length of the outer segments of these cells (See The Visual Pathway for details of the biosynthesis, maintenance and shedding of rod outer segment disc membranes under normal conditions).
Briefly, ROS disc proteins are synthesized in the inner photoreceptor segments and transported to the base of the outer segment where they are formed into discs. The older discs are pushed to the top of the outer photoreceptor segments where they are eventually shed and phagocytized by the RPE.
In the RCS rat The process of phagocytosis by the retinal pigment epithelium seems to fail
In the early stages of the development of the visual cell outer segments. The uniform stack-like arrangement of discs in the outer segmnents is disrupted and disc debris starts to accumulate between the tips of the photoreceptor outer segments and the pigment epithelial
cell layer.
Pigmented cells from the pigment epithelium invade the layer of debris and starts to
remove the debris and the degenerated photoreceptor cells. This process might coincide with the first appearance of the black spots or
"bone spicules" on the retinas of patients afflicted with Retinitis pigmentosa.
AS more photoreceptor cells degenerate, the layer of debris increases in thickness and the pigmented cells entering
the area of the photoreceptor layer increases presumably to remove
the degenerated cells.
The retina becomes thinner and more black spots appear on the
fundus as observed by the ophthalmoscope. These spots become larger as the black spots coalesce. Thus, the migration of retinal pigment cells into the area of the degenerated human photoreceptor cells might explain the appearance of the black spots on the fundus of the RP patients.
Why is there a failure of the phagocytic process in RP?
Back to TOP
The causes of retinal degeneration in retinitis pigmentosa are not yet known. Defects in genes encoding for proteins and enzymes of the photoreceptor outer segments and those involved in the transport of vitamin A from and to the outer segments of the photoreceptors have been found in patients presenting with RP and RP-like symptoms.
Although the list of gene defects
associated with RP is rapidly expanding, it is not however, known how any of the
mutations lead to photoreceptor dysfunction and death.
Three different suggestions, based on experimental results, were presented to explain degeneration in retinal dystrophies (4.)
A. Mutations in genes encoding the Structural Proteins of rod outer Segments
Mutations in the opsin and peripherin/RDS genes have been reported in association with changes in the ROS of some animals and in patients presenting with Autosomal Dominant Retinitis pigmentosa.
Mutations in the Opsin Molecule:
Investigations using molecular genetic techniques, have found more than 100 mutations or small deletions in the opsin molecule of patients presenting with autosomal dominant RP. These mutations cause addition or deletions in the opsin amino acid chain ranging from position 17 in the amino terminal of rhodopsin to the penultimate
position 347 at the carboxyl terminus.
Mutations in the opsin molecule might affect the normal folding of the molecule, or its capacity to bind vitamin A.
The large number of mutations in the rhodopsin gene might reflect some of the extensive clinical variability among patients with autosomal dominant RP.
Rhodopsin mutations that cause Autosomal Recessive RP are very infrequent:
However,
a patient, from a consanguineous marriage, was found to be homozygous for a mutation in the rhodopsin gene that caused a premature termination of the
transcription of the gene. This resulted in the complete absence of functional protein.(5.)
Another recessive mutation was observed in
a different family from consangnineous origin, segregating in a
a mutation that changes a single codon in the rhodopsin gene. The authors concluded that the resulting rhodopsin was unable to activate transducin.(4.)
Mutations in Peripherin/Retinal Degeneration Slow (Peripherin/RDS) gene
Mutations in the peripherin/RDS protein was originally found in the RDS (Retinal Degeneration Slow) mouse.
The mutation in the pripherin/RDS protein was found to be caused by a large insertion in the peripherin/RDS gene which interferes with the transcription and translational processes so that no protein will be synthesized. As a result, mice homozygous for the mutation
completely lack peripherin/RDS and develop
no outer segments.
On the other hand heterozygous mice, which have a reduced concentration of
peripherin/RDS, outer segments were present in normal numbers but were shortened and
displayed structural disorganization. The retinas of these mice degenerated slowly with time. These finding clearly demonstrating the importance of the peripherin/RDS protein in the development and structural integrity of the ROS
In humans, there is extensive phenotypic variation among individuals heterozygous for the peripherin/RDS gene . Affected individuals display either a autosomal dominant RP pattern,
dystrophy of the RPE, or cone and cone-rod
dystrophies. These phenotypes all
result from the degeneration of photoreceptors, but
differ in the extent of rod and/or cone involvement.
It is possible that insufficient periphein/RDS leads to instability of the ROS and affects the continuous internalization and degradation of outer segment disc membranes by the RPE. This poses a substantial burden on the metabolism of the RPE cell leading to their atrophy, and the degeneration of overlying photoreceptor
cells.
Mutations in the Rod Outer Segment protein 1 (ROM 1)
The ROM1 protein is present in rods and absent from cones. As ROM 1 normally interacts with peripherin/RDS it would be expected that mutations in
the ROM 1 gene would cause phenotypes similar
to those observed for peripherin/RDS mutations.
However, gene mutations alone in the ROM1 protein did not seem to cause disease
Digenic Retinitis Pigmentosa
Exceptions to the reports which indicate that mutations in the ROM 1 gene does not cause retinal
degeneration are found in cases in which mutations in bothe the peripherin/RDS and the ROM 1
genes occur simultaneously to form the "Digenic RP".
In all cases of digenic RP the mutations in the peripherin/RDS gene occurred at one locus whereas
three different ROM1 mutations were identified
in the families TESTED. Mutations in both the genes for the peripherin/RDS
and one of these ROM1 mutations must be present in order to lead to retinal disease.
The RP phenotype observed in digenic RP probably results from defective development or
shedding of the outer segments.
Patients of digenic RP present as psuedodominant RP. Carriers of the digenic disease have a 25% chance of transmitting the mutant peripherin/RDS and the ROM1 gene to his/her offspring.
How do mutations in the structural proteins of the rod outer segments lead to photoreceptor degeneration in RP?
It was suggested that the mutations in the genes encoding the outer segment structural proteins opsin, peripherin/RDS and ROM1would result in:
It was explained these accumulations of these proteins would disrupt the formation of the outer ROD rod outer segment discs thus leading to the shortening and disruption of the structure, of the rod outer segments.
On the other hand, mutations that prevent the phagotosis of outer segmemt discs
by the RPE, would cause accumulation of ROS disc debris between the photoreceptor cells and the RPE. This
would interfere with the normal passage of metabolites betweenn the outer segments and the RPE leading to the disruption the normal functioning of both the RPE and thephotoreceptor cells.
Back to TOP
B. The Phototransduction Cascade
Note: If you are not familiar with the details of the Phototransduction Cascade, please visit:
The Visual Pathway
Presented below is a simplified flowchart of the Phototransduction Cascade including the proteins and enzymes in this reaction
The visual pigment, rhodopsin, makes up 85 % of the proteins of the photoreceptor outer segments. It consists of two different molecules:
- Opsin, a single polypeptide chain consisting of 348 amino acids
- 11-cis retinal: a special form of vitamin A that is combined to Amino acid number 296 in the opsin polypeptide chain.
When light enters the eye it is absorbed by rhodopsin leading to a a conformational change in the structure of rhodopsin and the isomerization of its vitamin a from 11-cis retinal to all-trans retinal.
The altered form of rhodopsin, Metarhodopsin II, is a chemically active protein which reacts and activates another protein, transducin (a protein which consists of 3 polypeptides: alpha, beta and gamma polypeptides) combined with the dinucleotide, GDP. Activation of transducin causes the separation of the transducin alpha polypeptide from the beta and gamma chains and the exchange of the GDP bound to the alpha chain for GTP.
Transducin alpha.GTP in turn activates the enzyme cyclic guanosine monophosphate phosphdiesterase, a protein which consists of 4 polypeptides: alpha, beta and two gamma polypeptides.
This activation leads to the release of the two gamma polypeptides from the PDE and the hydrolysis of the GTP which is bound to the alpha polypeptide of transducin, to GDP. This releases the transducin-alpha.GDP to recombine with transducin-beta-gamma polypeptides.
The activated PDE then hydrolyzes cyclic guanosine monophosphate (cGMP).
cGMP plays a very important role in vision.
Under normal dark conditions of illumination, cGMP keeps certain channels called "cGMP-gated channels", located in the outer segment cell membranes open. These channels allow sodium and calcium ions to enter the photoreceptor cell.
Sodium ions are costantly removed from the outer segments by travelling to the inner segments where they are extruded by a sodium-potassium-activated enzye. Some sodium are also removed from the outer segment by exchange with a calcium/sodium exchanger located in the outer segment cell membrane.
Calcium ions are constantly removed from the outer segments by the sodium/calcium ion exchanger mentioned above.
Exposure to light, causes the activation of cGMP-PDE and the hydrolysis of cGMP. This leads to the closure of the cGMP-gated channels thus blocking the entrance of sodium and calcium ions into the ROS.
Calcium however keeps going out of the outer segments via the sodium/calcium exchanger. This leads to a decrease in the concentration of calcium in the outer segments, a condition which activates the enzyme, guanylate cyclase whose activity leads to the formation of cGMP, thus increasing the concentration of cGMP within the photoreceptor outer segments and opening the cGMP-gated channels.
The central role of the Phototransduction Cascade in vision and its location in the outer segments, would make a mutation in one of the genes coding the the proteins, including the enzymes, involved in the phototransduction cascade prime suspects in the development of Retinitis pigmentosa.
A defect in any one of these proteins or enzymes would cause adisruption of the phototransduction cascade and lead to similar phenotypic results.(see Ref.4.)
1. Mutation in the opsin gene
The first protein in the cascade is Rhodopsin (opsin + 11-cis-retinal).
Mutations in the opsin Gene are usually found in patients with autosomal dominant retinitis pigmentosa. However, reports of Autosomal
Recessive RP was reported to have a mutation in the opsin gene which produce a markedly truncated odopsin molecules that would be functionally
inactive because of the absence of the sixth and seventh transmembrane domains, including
the 11 -cis-retinal attachment site.
2. Mutations in cGMP Phosphodiesterase
Some patients suffering from autosomal recessive RP were found to have mutations in the genes encoding the alpha- and beta-sub-
units of cGMP PDE. These mutations would cause inactivity of the cGMP PDE. thus leading to the accumulation of high concentrations of cGMP in the ROS and the permanent opening of the cGMP gated cation channels.
These patients are night blind from birth
and suffer from a progressive constriction of the visual field.
A similar mutation in the gene encoding the beta subunit of cGMP PDE of the rd/rd mouse (retinal degeneration mouse) , a phenotype with retinal degeneration similar to
that in humans is observed. In mice homozygous for the gene demonstrate severe degeneration of photoreceptor cells which is directly or indirectly triggered by the
rise in cGMP concentration.
3.Mutations in the cGMP-gated Cation Channel
The rod cGMP-gated cation channel is composed of three subunits (alpha beta and gamma). Five different
mutations in the gene encoding the alpha-subunit have been reported in patients with autosomal recessive RP These mutations were found to either abolish the synthesis of the cGMP-gated cation channel protein or result in the synthesis of an abnormal protein.
Experiments with cultured transfected cells suggests that the mutant channel proteins fail to reach the plasma
membrane and are trapped within the cell.
The absence of the cGMP-gated cation channels in the the rod plasma membrane will prevent sodium and calcium ions from entering the ROS thus leading to abnormally low sodium and calcium concentration in the cell.
Low calcium concentration in the ROS will activate retinal rod guanylate cyclase, and
lead to a high rate of cGMP synthesis and its accumulation in the ROS. This would resemble the condition found in the mutations for cGMP
PDE where the cGMP PDE is inactive leading to an accumulation of cGMP in the ROS.
C. Metabolism of Vitamin A in the Eye
Please refer to: The Visual Pathway for more details about the metabolism of Vitamin A in the Eye.
The outer segments, of course, contain millions of rhodopsin molecules but, to keep the process of vision going, two processes must occur:
1. the active Metarhodopin II must be reined-in and converted to rhodopsin in order to receive another photon of light.
This is accomplished by phosphorylation and reaction with the protein Arrestin to form Arrestin- Metarhodopsin II-phosphate. This reaction is followed by the release of all-trans retinal and opsin-phosphate from the arrestin-Metarhodopsin-phosphate complex.
Opsin-phosphate is dephosphorylated by opsin phosphatase to prepare it for combination with 11-cis retinal.
2. all-trans retinal must be re-isomerized to 11-cis retinal in order to combine with opsin to form rhodopsin.
This is accomplished by transporting the released all-trans retinal (see above) to the RPE whwer it is re-isomerized to 11-cis retinal and sent back to the ROS for recombination with opsin.
Vitamin A deficiency resulting from malnutrition or metabolic
disorders results in a phenotype similar to RP.
Characteristically, symptoms of vitamin A deficiency include:
- night blindness
- abnormalities in the electroretinogram
- abnormalities in the visual field
- white spots appear at the level of the RPE. This contrasts with the black spots which appear on the fundus in RP
Lasting retinol (vitamin A) deficiency ultimately
leads to photoreceptor degeneration in both humans and animals.
Mmutations in two protins located in the pigment epithelium, have been found in patients with equal rod and cone degeneration,
These patients exhibited deposits in the retinal pigment epithelium similar to the deposits found in the eyes of patients with vitamin A deficiency.
1. Cellular Retinaldehyde-Binding Protein (CRalBP)
The CRalBP is found in the RPE It acts as a substrate-carrier protein for 11-cis-retinol and 11-cis-retinal, thereby modulating the interaction between this protein and visual cycle enzymes.
Patients carrying the mutant gene express:
- atypical Autosomal Recessive RP with early onset
of night blindness, macular degeneration, and visual
loss progressing toward legal blindness in the third
decade of life.
- Numerous white dots scattered over
the entire fundus, similar to the phenotype resulting
from retinol (vitamin A) deficiency.
This mutation probably disturbs or even completely blocks the regeneration of 11-cis -retinal and hence the regeneration of rhodopsin.
2. Retinal Pigment Epithelium Protein 65 (RPE65)
RPE65 is a protein that is exclusively found in the RPE and makes up about 10% of its protein content. Its exact function is not known, although it is supposed to be part of the metabolism of vitamin A because it is associated with the plasma retinol-binding protein, 11-cis -retinol dehydrogenase, and a third unknown protein.
Reports of mutations in the RPE65 gene resulting in an autosomal recessive retinal degeneration have been reported in a number of families. Phenotypes varied from severe, early-onset RP to the syndrome of Leber's congenital amaurosis.
All of these mutations are likely to result in a partial or complete loss of function of the RPE65 protein, thus blocking the regeneration of 11-cis retinal and the formation of rhodopsin, which is required for visual function.
Back to Top
To-date only some rare forms of retinitis
pigmentosa associated with systemic neurological disease are now amenable to treatment, namely (1a.):
1. Bassen-Kornzweig disease: is an autosomal recessive disorder
characterized by a defect in the ability to secrete certain lipoproteins and a marked decrease in The absorption and transport of cholesterol, fats, and fat-soluble vitamins from the small intestine to the plasma and lymph. As a result, serum and, subsequently, tissues levels of cholesterol and trigylcerides are low.
In addition to systemic neurogical disorders starting in childhood, patients report decreased night
vision and show the fundus changes typical of retinitis pigmentosa. Deficiencies of vitamin A and vitamin E are thought to be causative of the
retinal and neurological degeneration.
It was reported that treatment with large doses of vitamin A in the early stages of the disease can result in restoration of retinal function within 24 hours. But a combination therapy of vitamin A and vitamin E was found to be required to slow the course of retinal degeneration over the long term. Advanced cases are not amenable to
treatment, as a result of the large loss of photoreceptor cells. The neurological and
muscular sequelae are also slowed with vitamin supplementation.
2. Friedreich-like ataxia with retinitis pigmentosa: patients showing symptoms of this disease present with a sharp decrease in serum vitamin E level. and show a
thymine-to-guanine transversion at nucleotide position 303 in the alpha-TTP
cDNA.
Patients with this disease are treated with daily doses of vitamin E leading to normalized serum vitamin E levels, and the stabilization of visual and neurological signs and symptoms.
3.classic Refsum's disease):
In this disease, mutations in the gene encoding a peroxisomal enzyme phytanoyl-CoA
alpha-hydroxylase result in the inability to alpha-oxidize phytanic acid. Thus leading to the accummulation of the acid in many tissues.
The onset of the disease is variable, with some patients
presenting in early childhood and others in the second or third decade,
with neurological symptoms occurring prior to the onset of visual symptoms.
Treatment involves:
- lowering phytanic acid levels in
stages by restricting High sources of phytol and chlorophyll,
- a carefully balanced caloric intake to prevent the liberation of phytanic acid stores from body fat into other tissues.
- limiting the Daily consumption of fat to reduce the intake of phytanic acid
- daily supplementation of essential fatty acids and vitamin E.
- In severe cases, blood dialysis to reduce phytamic acid has been found to be helpful.
small amounts of chlorophyll-containing foods may be returned to the diet after serum levels have been reduced to normal levels.
Favorable response to treatment has been observed in patients treated with the above mentioned regimen for four years, with stabilization of the retinal degeneration and appreciable improvement of neurological and dermatological
disease.
To date, there is no effective treatment to influence the eventual outcome of common forms of RP. However, it has been reported that a daily dose of 15,000 units of retinyl palmitate (Vitamin A) slows the course of the disease in patients suffering from RP and Usher Syndrome. Vitamin E, on the other hand, may have a detrimental effect on retinal degeneration ( 13 )
Experiments with mouse models of autosomal dominant RP exhibiting a mutation in the rhodopsin gene at the threonine-17 : methionine in the opsin gene showed that vitamin A administration reduced the rate of decline of the electroretinogram and protected against photoreceptor rod outer segment loss.(12.)
On the other hand, vitamin A palmitate had no effect on the decline of the electroretinogram of transgenic mouse models of autosomal dominant RP carrying the opsin gene mutation in the proline-347 : serine(13), thus suggesting that the vitamin is selective in its effect.(12.)
, thus suggesting that not all of thecommon forms of RP will respond to treatment with vitamin A palmitate.
During the past decades, numerous experiments on humans, and animals carrying mutations analogous to those in human RP, have been performed to better understand the degeneration and cell loss in RP.
Among the various procedures used were:
Retinal Transplantation
Micro-aggregate suspension or intact sheets of human fetal retina were introduced between the neurosensory retina and the retinal pigment epithelium of human subjects.
The results indicated, at best and depending on the age of the patient, either subjective or objective improved light sensitivity during the initial period of the transplantation. This "improvement" disappeared 3 to 13 months later (6,7.).
Modification of mutant gene products
Proteins are synthesized according to a code that is copied from genes by a molecule called messenger ribonucleic acid (mRNA). The latter is synthesized with additional nucleotides that are later trimmed to normal size by enzymes, called ribozymes.
A mutation in a gene for a particular protein would cause the synthesis of an abnormal mRNA which cannot be trimmed to size by ribozymes thus resulting in the synthesis of an abnormal (mutant) protein.
Ribozymes engineered to cleave mutant mRNA to an mRNA that would produce a normal opsin molecule, were delivered to the retinas of transgenic rat model of autosomal dominant RP. The results indicated a considerable decrease in the rate of photoreceptor degeneration in these rats for at least three months post-operatively.(8.,9.)
It thus appears that delivery of ribozymes to the photoreceptors, has broad potential for more applied therapeutic purposes especially since efficient mutation-independent delivery systems targeting rhodopsin and peripherin have been developnt. This prediction is encouraged by the availability of animal models ranging from mice to pigs which are being used as experimental models.
The application of growth factors and/or pharmacological agents.
Repeated injection of the human Ciliary Neutrophic Factor (CNTF) analogue, Axokine, into the intravitreal space of cats suffering from the autosomal dominant model of rod-cone dystrophy,
delays photoreceptor loss in this hereditary retinal
degenerative disease(10.).
The rds/rds mouse exhibits a mutation in the peripherin/RDS gene, which is linked to many forms of dominantly inherited retinal degenerations in humans. Injection of CNTF ribozyme into the eyes of the rds/rds mouse, reduces photoreceptor loss, causes a significant increase in the length ofphotoreceptor segments, and results in a redistribution and an increase in the retinal content of the photopigment rhodopsin.(11.)
These effects are accompanied by a significant increase in the amplitude of the a- and b-waves of the
electroretinogram indicating an improved response of the retina to light.(11.)
The above results would suggest that delivery of the CNTF ribozyme could potentially be useful in the treatment of some forms of retinal degeneration
Calcium Channel Blockers
rd/rd mice, are one of the animal models of autosomal recessive RP. They exhibit a deficiency in cGMP phosphodiesterase which leads to a large increase in the level of cGMP in photoreceptors. This increase is thought to lead to photoreceptor death, as a result of the continuous opening of the cGMP-gated channels.
Administration of the calcium-channel blocker, Diltiazem (usually used for the treatment of hypertension), to rd/rd mice, slowed the loss of photoreceptor cells in these mice.(15.)
Calcium-ion channel blockers have not yet been tried on human subjects suffering from autosomal recessive RP.
Back to Top
In his 1993 Friedenwald Award Lecture (2.) ( a prestigius award for investigators in the field of Ophthalmology) , Elliot Berson, a leading investigator in the field of retinal degeneration, concluded that:
"Much work remains to be done. It still is my belief,
as it was 20 years ago, that coordinated progams of
clinical and basic research offer the best hope of
finding treatments for patients with this group of diseases".
Both clinical and basic research on the causes of retinal degeneration and for finding therapies for the disease is ongoing. The development of new technics and the availability of animal models or retinal degeneration bode well for finding methods of amelioration or even treatment or retinal degenerations.
Back to Top
I am indebted to Mme Helene Dussiaume for invaluable assistance in searching for references and making them available to me.
Dr. Gilles Desroches, Ottawa, Ontario and and Dr. S. Coupland, Ottawa Eye Institute, Ottawa University, Ottawa, Ontario, Contributed valuable illustrations for this Page.
1. Massof, R., Finkelstein, D., and Boughman,
J. A.: Birth Defects, Orig. Artic. Ser. 18(6), 161 - 166, 1982
1a. Grant, Calvin A. M.D.; Berson, Eliot L. M.D.
Int Ophthalmol Clin, Volume 41(1).January 2001.103-110
2. Berson, EL: Invest. Ophthalmol. Vis. Sci. 34: 1659, 1993
3. Kajiwara, K., Berson, EL and Dryja, TP: Science 264: 1604, 1994
4. van Soest, S. et al.: Surv. Ophthalmol. 43:321, 1999
5.
Rosenfeld, JR, Cowley, GS, McGee, TL, et al: Nat. Genet. 1:209-213, 1992
6.
Radtke, ND, Aramant, RB, Seiler, M, Petry, HM:
Am. J. Ophthalmol. : 128 (3): 384-387, 1999
7. Ali, RR, Sarra, GM, Stephens, C,: Nat. Genet. 25 (3): 306-310, 2000
8. [published erratum appears in Nat Med 1998 Sep;4(9): 1081 ]
Lewin, AS, Drenser, KA, Hauswirth, WW:
Nat. Med. 4 (8): 967 - 971, 1998
9.Kumaramanickavel, G, Maw, M, Denton, MJ, et al.: Nat. Genet. 8: 10 - 11, 1994
10.
Chong, NH, Alexander, RA, Waters, L, et al: Invest. Ophthalmol. Vis. Sci. 40 (6): 1298 - 1305, 1999
11. Cayouette, M, Behn, D, Sendtner, M, et al. : J. Neurosci. 18 (22): 9282-9293, 1998
12.
Li, T, Sandberg, MA, Pawlyk, BS,: Proc. Natl. Acad. Sci. U.S.A. 1998 : 95 (20): 11933 -11938, 1998
13.Berson, EL, Rosner, B, Sandberg, MA, et al.: Arch. Ophthalmol. 111: 761 - 772, 1993
14.Sibulesky, L, Hayes, KC, Pronczuk, A et al.: Am. J.
Clin. Nutr. 69: 656 - 663, 1999
15.Frasson, M, Sahel, JA, Fabre, M, et al.
Nature Med. 5: 1183 - 1187, 1999
This page is still under construction and will be updated regularly. Please visit often and send me your
comments and suggestions:
HERE
P.S. I will be grateful to you if you can provide me with any human material to illustrate the various aspects of Retinitis pigmentosa.
Updated Thursday, September 25, 2003
Back to Top
 Click Here to see details of the image
Click Here to see details of the image Click Here to see enlarged image
Click Here to see enlarged image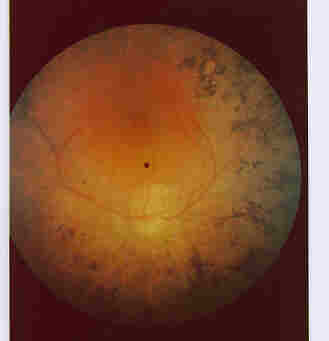 Click Here to see details of the image
Click Here to see details of the image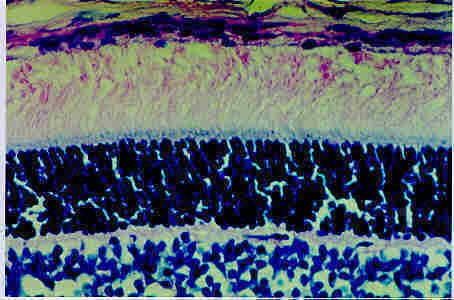 Click here to see details of the image.
Click here to see details of the image. Click here to see details of the image.
Click here to see details of the image.