Role of Huntingtin in cells
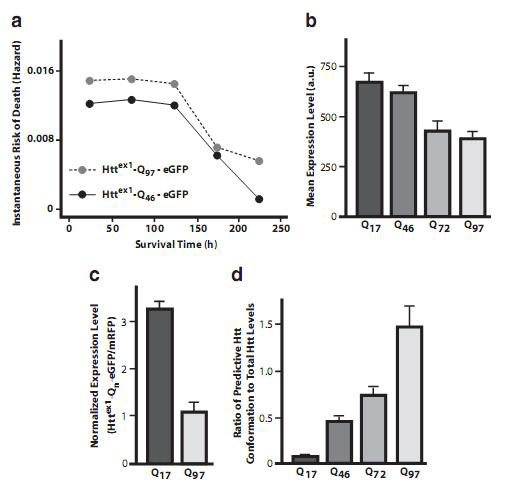 Figure 3. A summary of the relationship between glutamine length, expression levels, and disease severity described by risk of death. Taken from source 4.
Figure 3. A summary of the relationship between glutamine length, expression levels, and disease severity described by risk of death. Taken from source 4.
The Role of htt
Surprisingly the function(s) of the normal version of Huntingtin protein in a cell remains elusive (2). A role in intracellular trafficking has been proposed for Huntingtin as the normal and mutant version interacts with β-tubulin (7). Mouse embryo’s lacking the Huntingtin genes altogether do not survive (1, 2). This may be a consequence of normal Huntingtin aiding in the proper localization of the spindle proteins dynein, which is critical in proper cellular division (5). Analysis of the motifs on the Huntingtin gene reveals protein interaction domains and proteolytic cleavage sites, hinting that Huntingtin’s may participate in several pathways in a cell (2). A better understanding of the molecular pathways that Huntingtin participates in is crucial in treating this disease as it will shed light on what the disease causing mutant version does and how this could lead to degeneration of cognitive abilities. Functional analysis of each of the proteins domains (fig. 2) and their contribution to cellular homeostasis can be done to give a deeper insight into what this protein may do in a cell.
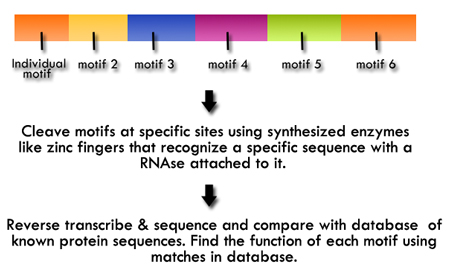
Figure 2. A method for investigating
each motif may contribute to protein
function.
Diseased state Huntingtin & quantitative traits.
The severity of this disease correlates well with the severity of the CUG trinucleotide expansion in exon one (4). In other words, the level of expansion of glutamine at the amino acid sequence level correlates to the severity of Huntington’s disease. Quantitative studies using fluorescence signals reveal that the bigger the mutation and hence the larger the protein, the less of it is expressed in cells (4). From the data in figure 3, the larger mutant forms seem to compensate for their low levels inside rat neuronal cells. Larger expansions of glutamine repeats seem to be positively related to increased risk of death and hence disease severity.
Since patients with Huntington’s disease would most likely have a different length of glutamine repeats and severity state, each patient must be treated differently. Personalized medicine would be an effective way at treating patients suffering from this disease as their level of severity would mostly differ from patient to patient. Even if there was an effective drug out there that could treat this disease and delay neurodegeneration, would the dosage be the same for every patient? Genetic counseling and screening of individual patients for their Huntingtin gene can reveal how severe their mutation is and allow for treatment design before onset of symptoms surface.