

 |
Genetics and Evolutionary AspectsHypothesis: SARS-associated Coronavirus is a separate genus from other Coronavirus
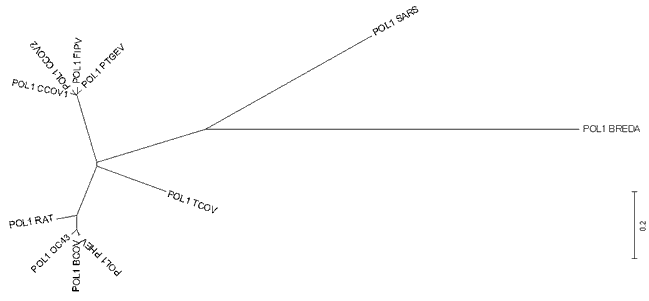
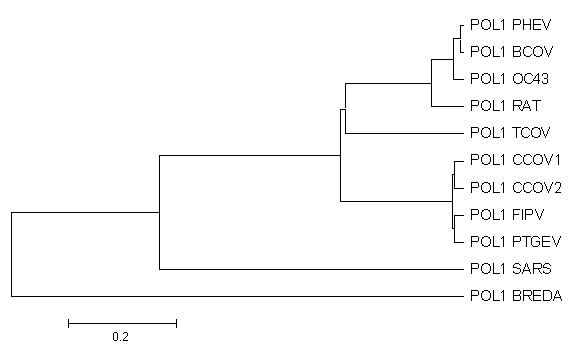
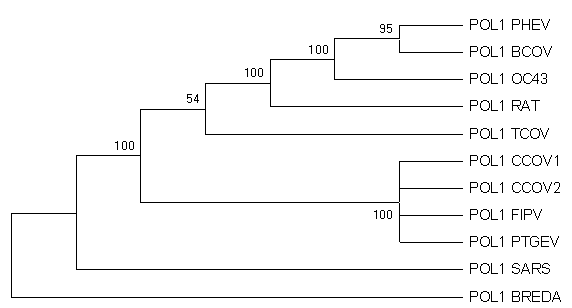
MethodsThe nucleotide sequence of Urbani-SARS coronavirus ORF1b RNA polymerase was obtained from the WHO Collaborative Networks3. The nucleotide sequence was translated to amino acid sequence (Figure 1). The translation was verified with BLASTX (translated BLAST search - nucleotide query to protein db). Nucleotide sequences of RNA-directed RNA polymerase from Canine coronavirus UWSMN-1 (gi|22774013); Porcine transmissible gastroenteritis virus (gi|4927036); Turkey coronavirus (gi|4927034); Rat sialodacryoadenitis coronavirus (gi|4927032); Human coronavirus (strain OC43) (gi|4927030); Porcine hemagglutinating encephalomyelitis virus (gi|4927028); Feline infectious peritonitis virus (gi|4927026); Canine coronavirus (gi|4927024); Bovine coronavirus (gi|4927022); and Breda torovirus (gi|4204756) were obtained from NCBI nucleotide database. The sequences were aligned using the program ClustalW 1.81 (ftp://ftp.ebi.ac.uk/pub/software/dos/clustalw) with default parameters (gap opening penalty=10, gap extension penalty=0.20). The alignments were then formatted to a MEGA format. The alignments were visualized in MEGA version 2.1 (Kumar et al. 2002). A preliminary phylogenetic tree was constructed with the following method in place: Unweighted Pair Group Method with Arithmetic mean (UPGMA), Kimura 2-parameter, and bootstrap with 100 replications. Tajima relative-rate tests are then performed between SARS-associated coronavirus, Bredavirus, and coronavirus from all three antigenic groups. Results and DiscussionsFigure 2a: Radiation Tree
Figure 2b: Traditional Rectangular View
Figure 2c: Bootstrap Consensus Tree
Table 1: Pairwise Distance between all viruses examined, using the Kimura 2-Parameter method
Average
Pairwise Distance between the Three Antigenic Groups and
SARS-associated Coronavirus:
Viruses within the coronavirus family separate into three antigenic groups as expected. However, the genetic distance between SARS and other coronaviruses is similar to the distance between SARS and Bredavirus but different from the distance between coronaviruses from all three antigenic groups (see the above table). This is something not found in other publications on SARS thus far. This, in conjunction with the UPGMA tree obtained, seems to suggest that the SARS-associated coronavirus diverged very early in the evolution from the ancestors of coronavirus, soon after the divergence of Bredavirus (a torovirus subfamily) from its common ancestor. Preliminary results from Tajima relative-rates test between SARS-associated coronavirus, Bredavirus, and coronavirus from all three antigenic groups suggest that the SARS-associated coronavirus mutated at the same rate as other coronaviruses. This decreases the likelihood that the SARS-associated coronavirus is mutating faster than expected. These results suggest that perhaps the new virus is a separate genus distinct from other known coronavirus. Next Step
|
Last Modified: