Research
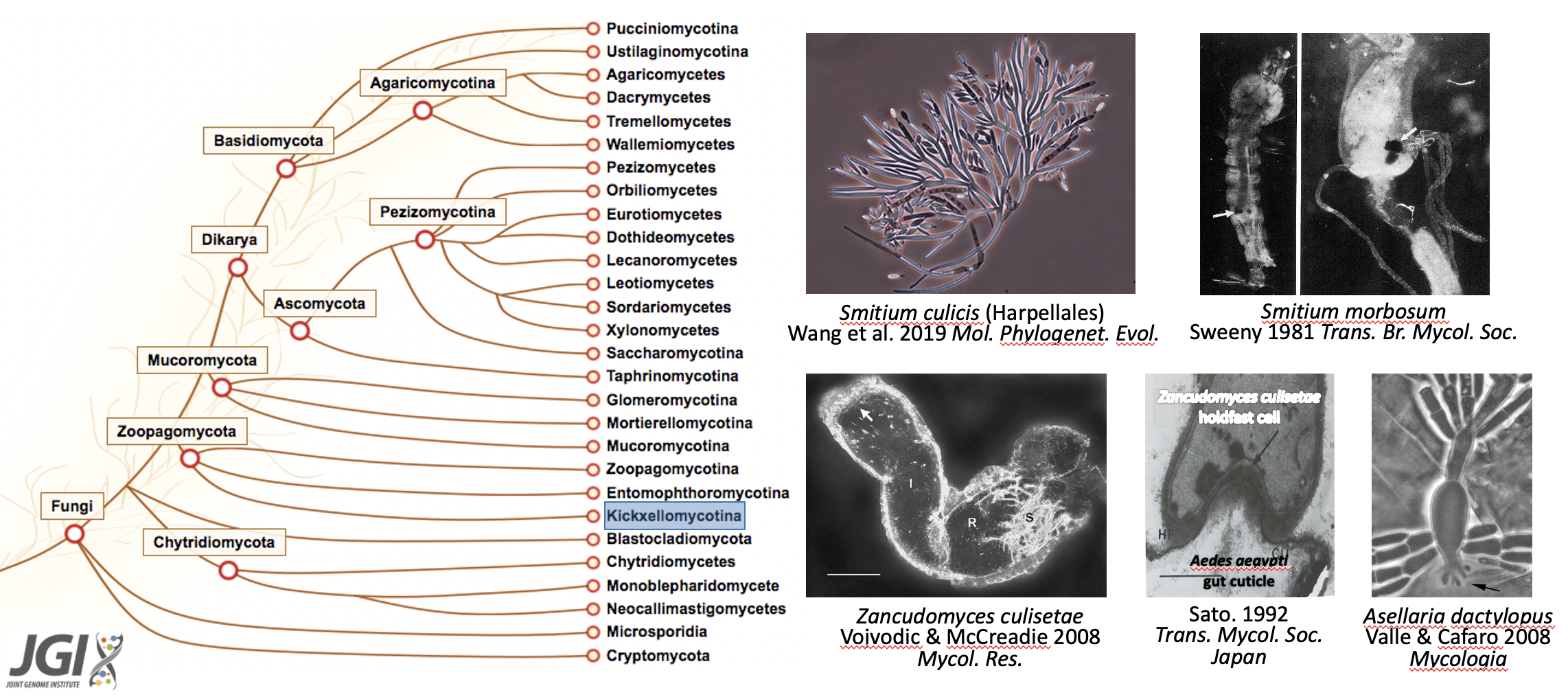
We study fungal ecology and evolution by integrating modern genomics with traditional taxonomic methods, with a specific focus on the early-diverging groups (e.g., zygomycetes, insect gut-dwelling fungi, anaerobic gut fungi). We use Harpellales fungi (symbionts) and insects (e.g., mosquitoes) as the model to study genomic interactions between gut fungal symbionts and their disease-bearing insect hosts.
Specific Topics:
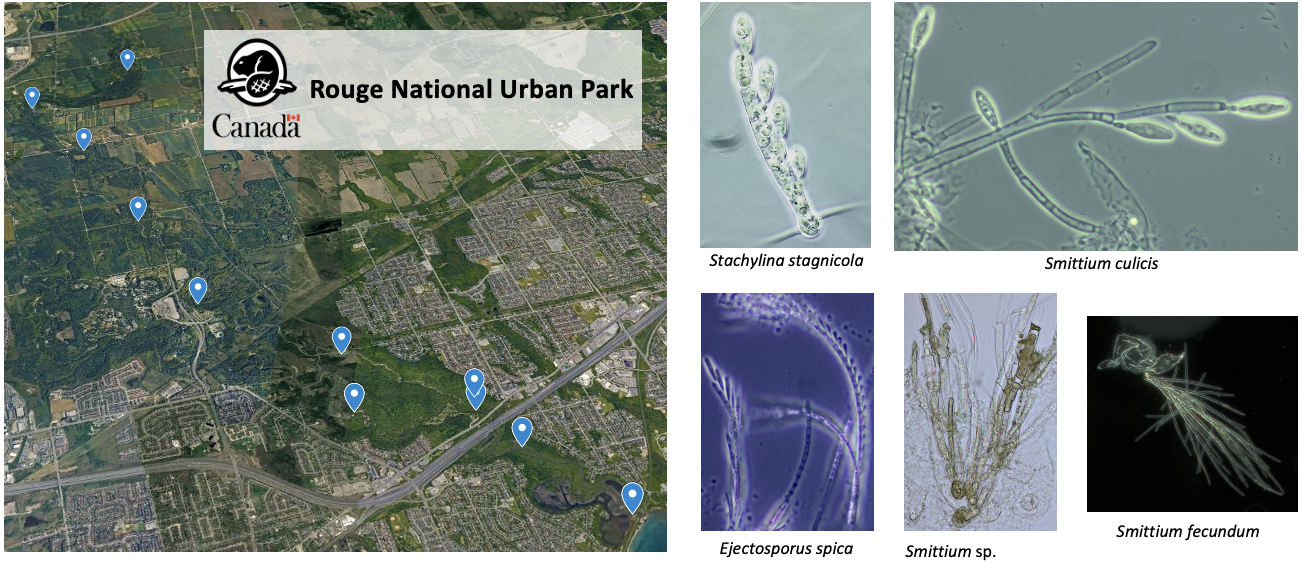
1) Fungal diversity in Ontario aquatic systems
Our lab dedicates to identify and collect fungal species residing in Ontario watersheds including the Rouge River in the Rouge National Urban Park close to our campus. We consistently train students at both undergraduate and graduate levels and a few spots are currently available. Interested students should contact the PI directly. Check Lehn et al. 2025 for more details.
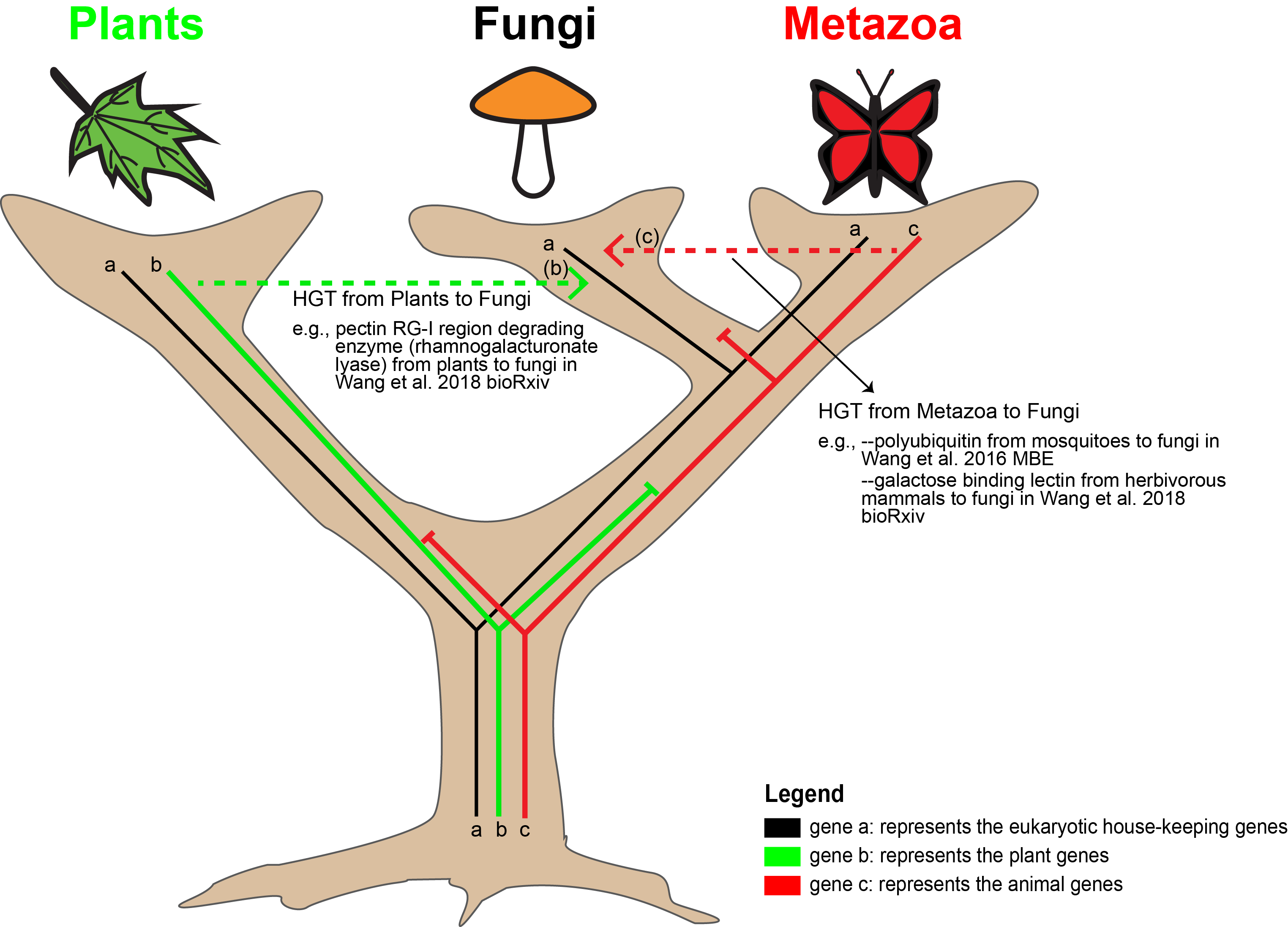
 2) Horizontal Gene Transfer (HGT) in Fungi
2) Horizontal Gene Transfer (HGT) in Fungi
HGT has now been known as a major factor to influence species evolution and adaptation. Although its mechanism has not been understood in eukaryotes, HGT has been reported from several lineages of fungi. A Python script has been developed to facilitate the HGT identification (https://github.com/YanWangTF/HGTfilter) with a special focus to identify host originated HGT in symbiotic fungi. One gene that encodes a mosquito-like polyubiquitin chain was successfully identified (Wang et al. 2016 MBE). Similarly, the tool also helped reveal more examples of HGT from animals and plants to another early-diverging fungal group, Neocallimastigomycota (Wang et al. 2019 mSystems).
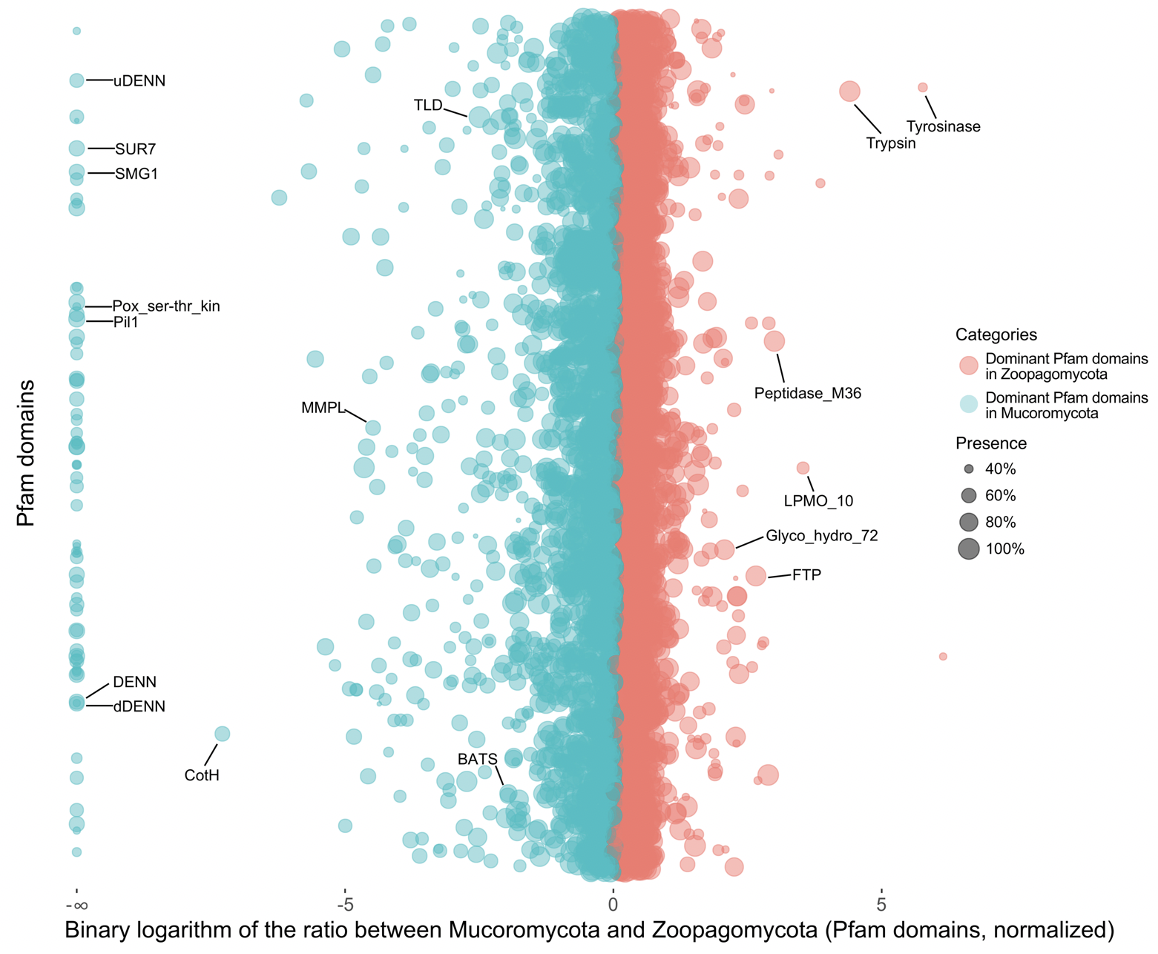
3) Symbiosis and Comparative Genomics
A classic question in evolutionary biology is how organisms form symbiotic relationships and how they co-evolve with each other. Our lab study the origin and molecular mechanisms of fungus-host associations. In a recent study (Wang et al. 2023 GBE), we applied comparative genomic approach and identified multiple protein family domains that may have contributed to the divergent evolution of zygomycete fungi with either plants or animals. In terms of the insect-associated group (Wang et al. 2018 mBio), we discovered a set of genetic tools, named Fungus-Insect Symbiotic Core Genes (FISCoG), which are shared among insect commensal and pathogenic fungi and are responsible for adhesion, hydrolysis, to host immune repression. In a separate study, a comparative genomic method was developed that helped reveal a unique set of protein family domains maintained by the mammal gut fungi (Neocallimastigomycota), but lost in the rest of fungal kingdom (Wang et al. 2019 mSystems). These elements were suggested to be important in explaining the exceptional ability to degrade plant biomass in the rumen for their herbivore hosts
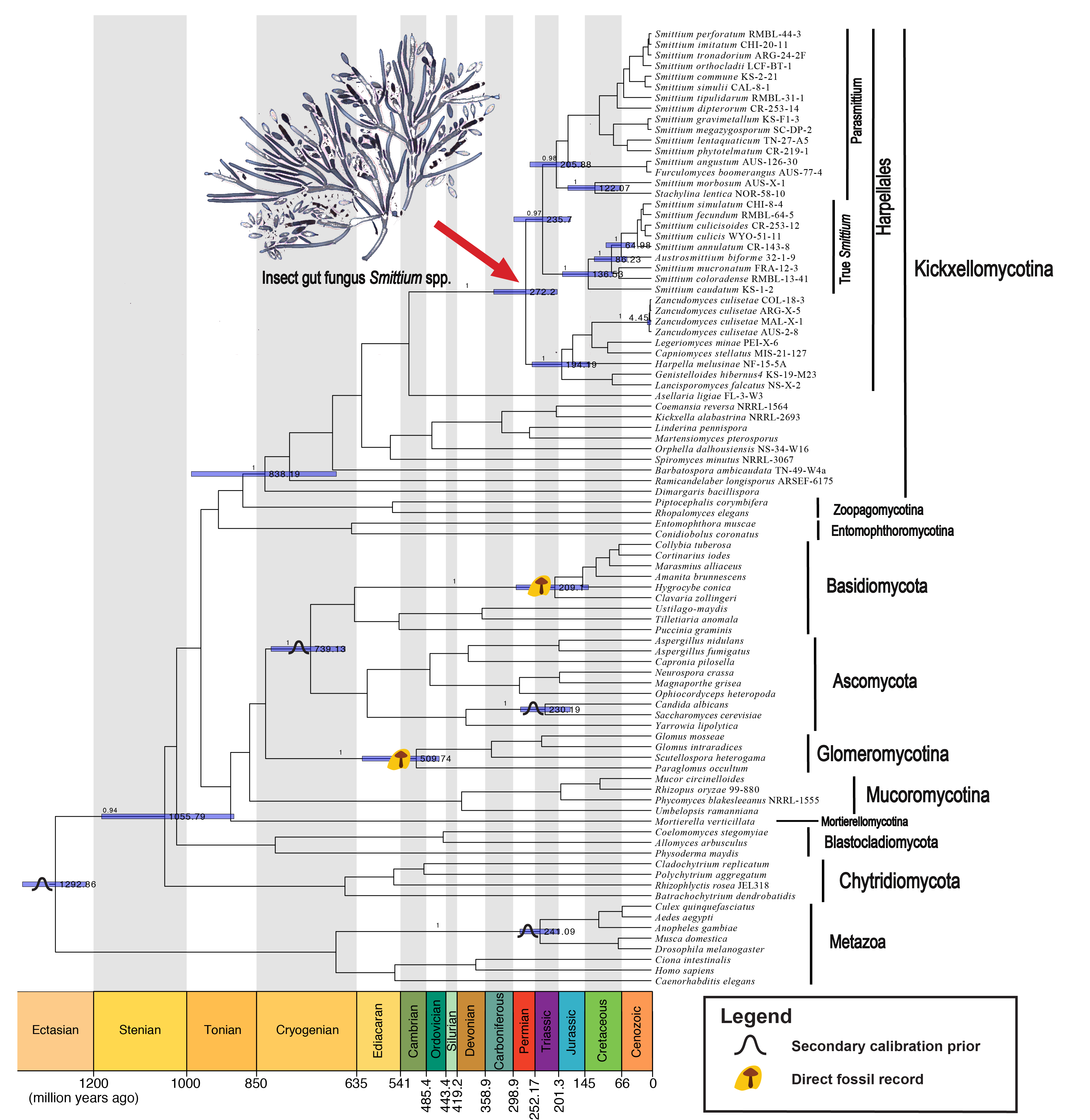
4) Fungal Phylogeny and Divergence Time Estimation
Phylogenetic reconstruction and divergence time estimations are informative in recovering the evolutionary kinships of various fungal organisms and deciphering how they have occupied the current territory. These approaches allow us to predict and investigate the roles of fungi in shaping the environment. Taking advantage of the development of molecular phylogenetic markers with increasing resolving powers, I have applied modern phylogenetic methods to establish a new fungal genus (Wang et al. 2013 Mycologia), delimitate species complexes (Wang et al. 2014 MPE), estimate divergence time of certain fungal clades (Wang et al. 2019 mSystems), and study the co-evolutionary relationship with symbiotic partners (Wang et al. 2019 MPE).